- + Merck Millipore
- + Thermo Fisher
- + IKA
- + 顯微鏡
- + 光度計
- + 成像系統
- + MACS Miltenyi
- + 均質機
- + 粒子計數器
- + 凍干機
- + 滅菌系統
- + 細胞
- + 微量移液工作站
- + 振蕩培養箱
- + 生物反應器
- + 切片機
- + 培養箱
- + 蠕動泵
- + 細胞破(粉)碎機
- + 轉印膜
- + 超濾管
- + Pellicon 超濾系統
- + 超低溫冰箱
- + 清洗機
- + 干燥機
- + 洗瓶機
- + 離心泵
- + 容積泵
- + 各種閥
- + 酶標儀
- + 洗衣板
- + 旋光儀
- + 折光儀
- + 行星球磨機
- + 振動篩
- + 基因導入儀
- + 手套系列
- + 接頭\\連接器
- + 培養板/培養瓶
- + 溫度控制系統
- + 制冷器
- + 存取系統
- + 軋蓋機
- + 細胞因子
- + 細胞分選儀
- + 生物安全柜
- + 滲透壓儀
- + 拉曼光譜儀
- + 電泳系統
- + 純水系統
- + 萃取儀
- + 譜新生物
- + TA 儀器
- + wako
第一期 || 淺談IVD行業公用系統監測和微生物檢測法規
更新時間:2019-12-03 瀏覽次數:2877
國慶長假已經結束~祝賀大家喜提2019年所有法定節假日,接下來又該投入忙碌的科研工作生活了!
小編在假期前的推文中留了一個“小尾巴”——“基于樣品檢測量多少的變更需要進行適用性檢查嗎?”
(點擊回顧→ “國慶堵”vs“濾膜堵”)
對于這個問題,我們的建議是:由于原來10ml的供試液較1ml的藥濃更高,如含抑菌成份則抑菌性更強,所以在方法適用性檢查上能包括濃度更低的供試液,所以建議只需走公司內部規定的變更-修改SOP和記錄流程即可,不需要重新進行驗證。
解決完“遺留問題”,接下來本文將以法規要求作為指導幫助大家梳理IVD行業中關于微生物監測的方法。
醫療器械生產質量管理規范(GMP)在2015年10月份生效,其中有三大附錄:《無菌醫療器械》、《植入性醫療器械》、《體外診斷試劑》。
下面我將整體分析IVD行業關于公用系統和檢測樣品方面的微生物控制。
?《醫療器械生產質量管理規范附錄體外診斷試劑》
2.2.3應當根據體外診斷試劑的生產過程控制,確定在相應級別的潔凈室(區)內進行生產的過程,避免生產中的污染。空氣潔凈級別不同的潔凈室(區)之間的靜壓差應當大于5帕,潔凈室(區)與室外大氣的靜壓差應大于10帕,并應當有指示壓差的裝置。相同級別潔凈室間的壓差梯度應當合理。
2.2.5陰性或陽性血清、質粒或血液制品等的處理操作,生產區域應當不低于10000級潔凈度級別,并應當與相鄰區域保持相對負壓。
2.2.11潔凈室(區)的溫度和相對濕度應當與產品生產工藝要求相適應。無特殊要求時,溫度應當控制在18~28℃,相對濕度控制在45%~65%。
2.2.19進行危險度二級及以上的病原體操作應當配備生物安全柜,空氣應當進行過濾處理后方可排出。應當對過濾器的性能進行定期檢查以保證其有效性。使用病原體類檢測試劑的陽性血清應當有相應的防護措施。
2.2.20對于特殊的高致病性病原體的采集、制備,應當按照有關部門頒布的行業標準,如人間傳染病微生物名錄、微生物和生物醫學實驗室生物安全通用準則、實驗室生物安全通用要求等相關規定,配備相應的生物安全設施。
2.3.1潔凈室(區)空氣凈化系統應當經過確認并保持連續運行,維持相應的潔凈度級別,并在一定周期后進行再確認。若停機后再次開啟空氣凈化系統,應當進行必要的測試或驗證,以確認仍能達到規定的潔凈度級別要求。
2.6.9應當制定潔凈室(區)的衛生管理文件,按照規定對潔凈室(區)進行清潔處理和消毒,并做好記錄。所用的消毒劑或消毒方法不得對設備、容器具、物料和產品造成污染。消毒劑品種應當定期更換,防止產生耐藥菌株。
2.7.2生產和檢驗用的菌毒種應當標明來源,驗收、儲存、保管、使用、銷毀應執行國家有關醫學微生物菌種保管的規定和病原微生物實驗室生物安全管理條例。應當建立生產用菌毒種的原始種子批、主代種子批和工作種子批系統。
以下為內容大綱,本文將從三個方面進行分析梳理,今天先從“潔凈度監測”方面展開。
?1、潔凈度監測
2、溫濕度和壓差監測
3、病原微生物實驗室
一、潔凈度監測
? 相關法規
《醫療器械生產質量管理規范》
《GB 50591-2010 潔凈室施工及驗收規范》
《GBT 16292-2010 醫藥工業潔凈室(區)懸浮粒子的測試方法》
《GBT 16293-2010 醫藥工業潔凈室(區)浮游菌的測試方法》
《GBT 16294-2010 醫藥工業潔凈室(區)沉降菌的測試方法》
《ISO 14644-1:2015 潔凈室和相關受控環境 第一部分:根據粒子濃度劃分空氣潔凈度等級》
《2010年藥品GMP指南:無菌藥品》
? 粒子監測
潔凈區參考《GB 50591-2010》的要求,按照《GBT 16292-2010》方法對粒子進行監測,級別為百級、萬級、十萬級,生產或檢測區域潔凈度不低于十萬級。
根據《GBT 16292-2010》描述,該法規是參照《ISO 14644-1》,現新版為2015版,建議按《ISO 14644-1:2015》執行,較《ISO 14644-1:1999》和《GBT 16292-2010》,其在取樣點數、采樣次數、zui小取樣量、結果判定、取樣管長度、粒子計算器符合要求均有所不同。
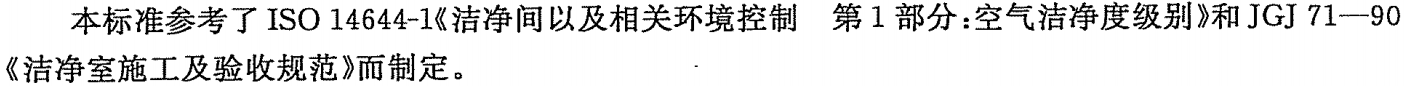
▲《GBT 16292-2010》
? 沉降菌和浮游菌監測
培養基和時間參考《GBT 16294-2010》,TSA在30-35℃培養不小于2天,SDA在20-25℃培養不小于5天;但zui少采樣點數目不應參考《GBT 16294-2010》,應參考《ISO 14644-1:2015》。


▲《GBT 16294-2010》
? 標準方面
參照醫療器械GMP,以下是沉降菌動靜態監測0.5小時的結果,而粒子數結果判定不需要計算95%UCL;
推薦使用Merck環境監測成品培養基和MAS-100 NT浮游菌采樣器


? 監測頻率
《醫療器械生產質量管理規范附錄體外診斷試劑》
2.3.1潔凈室(區)空氣凈化系統應當經過確認并保持連續運行,維持相應的潔凈度級別,并在一定周期后進行再確認。若停機后再次開啟空氣凈化系統,應當進行必要的測試或驗證,以確認仍能達到規定的潔凈度級別要求。
靜態監測要求參考《GB 50591-2010》,動態監測根據用戶風險評估制定;
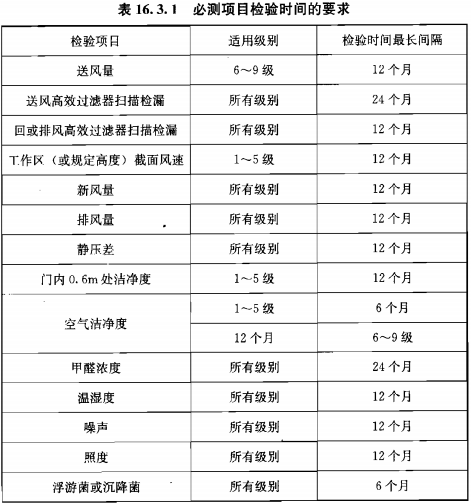
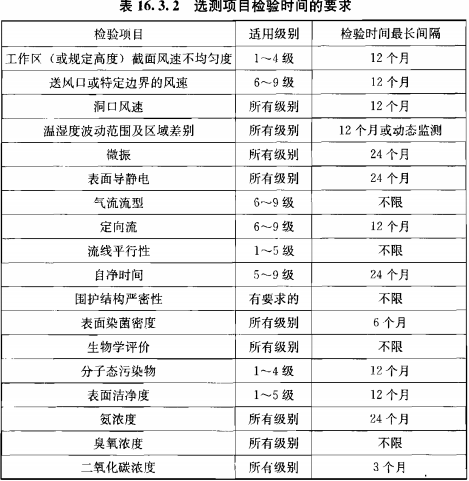
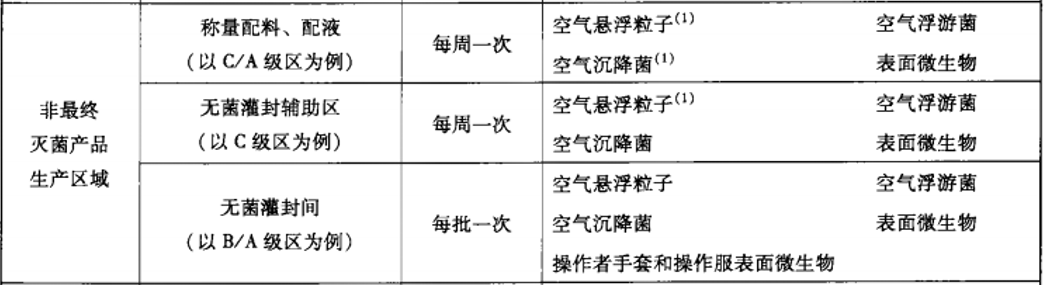
建議動態監測參考2010年藥品GMP指南:無菌藥品,百級每批生產,萬級、十萬級每周一次;
推薦使用TSI 9500系統便攜式粒子計數器,有28.3L、50L、100L型號,用于百級、萬級、十萬級潔凈區的塵埃粒子監測。內含多種標準《ISO14644-1:1999》、《EUGMP-ISO:1999》、《ISO14644-1:2015》、《EUGMP-ISO:2015》,適合制藥行業和醫療器械行業的使用。輸入”面積”、“潔凈等級”、“選擇標準”和“房間狀態”后自動生成“通道”、“取樣量”、“位點數”、“取樣時間”等的數據,其數據完整性符合FDA 21 CFR PART11、Part211和212要求;

以上就是“潔凈度監測”的分析內容,下期將圍繞“溫濕度和壓差監測”展開分析梳理,敬請期待!
- (上一篇):想除熱原?買它!!
- (下一篇):生物安全柜怎么用更安全?